什么是BSA
BSA(体分离分析)是一种QTL作图技术,用于鉴定影响感兴趣性状的基因组位点。该技术涉及形成两组以极其相反的表型进行个体杂交,或者将感兴趣的突变体杂交为野生型,然后通过从表型分布的尾部选择个体并进行池测序来创建两个群体,然后估计这两个群体的等位基因频率,如果基因组中包含因果位点的区域存在差异,则这两个人群之间会表现出差异。

什么是BSA-seq
随着人口分组技术的进步和 下一代测序(NGS) 已显著减少,整合 全基因组重测序 使用块状分离分析(BSA)已成为一种突出的方法。BSA与NGS的融合,称为BSA-seq,加快了关键性状紧密连锁标记的鉴定,从而增强了基因发现,提高了数量性状位点(QTL)作图的分辨率。值得注意的是,BSA-seq在构建实验群体方面节省了大量时间,特别是在精确定位数量性状位点(QTL)时,使其成为鉴定与数量性状相关的功能基因和位点的快速有效手段。总之,BSA-seq植根于群体分组和第二代测序技术的融合,能够识别与表型性状相关的单核苷酸多态性(SNP)位点。它在QTL的精确定位和功能靶基因的鉴定中得到了广泛的应用。
BSA-seq技术原理
BSA-seq技术解决了与在作物中创建近等基因系(NIL)相关的许多局限性。其原理涉及将一对表现出相反感兴趣特征的亲本系杂交。随后,从任何分离的后代群体(通常是F2代)中,选择20至50株表现出目标性状极端表型的单株。然后提取这些个体的DNA并等摩尔混合,从而形成两个不同的基因库。这两个基因库在感兴趣的性状方面应该有所不同,而所有其他基因组位点仍然是随机的。通过比较这两个库确定的多态性标记可能与感兴趣的功能基因或数量性状位点(QTL)有关。
BSA实验流程图
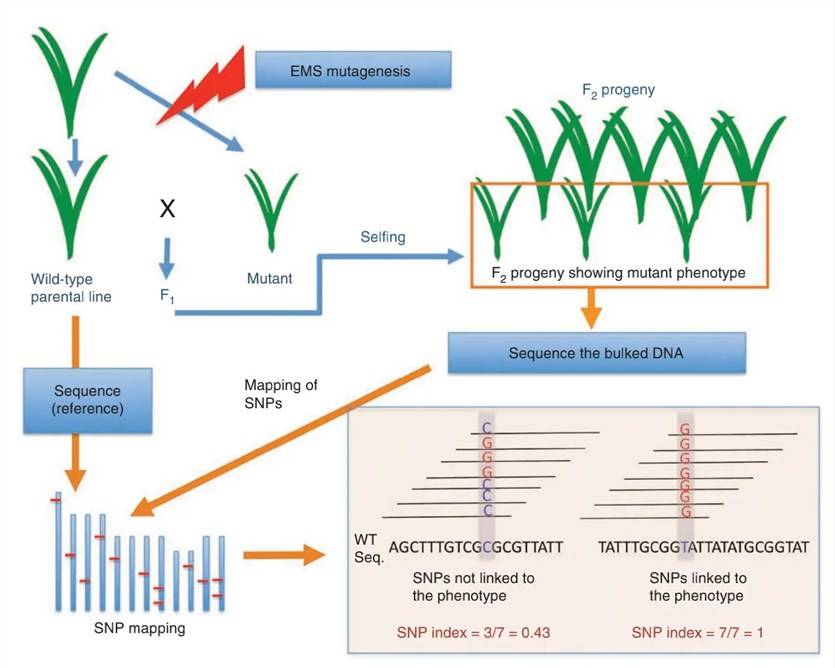 《自然生物技术》,2011年,Akira Abe等人。
《自然生物技术》,2011年,Akira Abe等人。
主要优势和特点
- 实验周期短
- 精确的测绘结果
- 成本效益高
常用方法
- QTL-seq:天然突变基因的定位
- Mutmap:人工突变基因的定位
服务规范
样品要求
|
|
测序服务
|
|
 |
生物信息学分析 我们提供定制的生物信息学分析,包括:
|
BSA-seq数据分析
To identify functional gene loci, a comparison at the SNP level between the two pooled and sequenced populations is essential. The most commonly employed method for this purpose is the SNP-index approach. Its underlying principle involves the statistical analysis of bases at each nucleotide position using the sequencing reads. A reference parent or an existing reference genome is typically selected as a reference. The counts of reads in the offspring pool that match or differ from the reference at a specific nucleotide position are tallied, and the ratio of differing reads to the total reads at that position is calculated, yielding the SNP-index. The SNP-index is determined within a sliding window, usually using a 1 Mb window size with a 10 kb increment for each slide. Additionally, Δ(SNP-index) is used to measure the difference in SNP indices between the two gene pools, effectively capturing variations at individual loci. The results are typically visualized in a Manhattan plot, as depicted below, where the horizontal axis represents chromosomal positions, the vertical axis of the SNP-index plot indicates the computed read ratios for each SNP position, and the vertical axis of the Δ(SNP-index) plot represents the difference in SNP-indices between the two pooled samples. The larger the absolute value of Δ(SNP-index), the more significantly associated a locus is with the extreme trait.
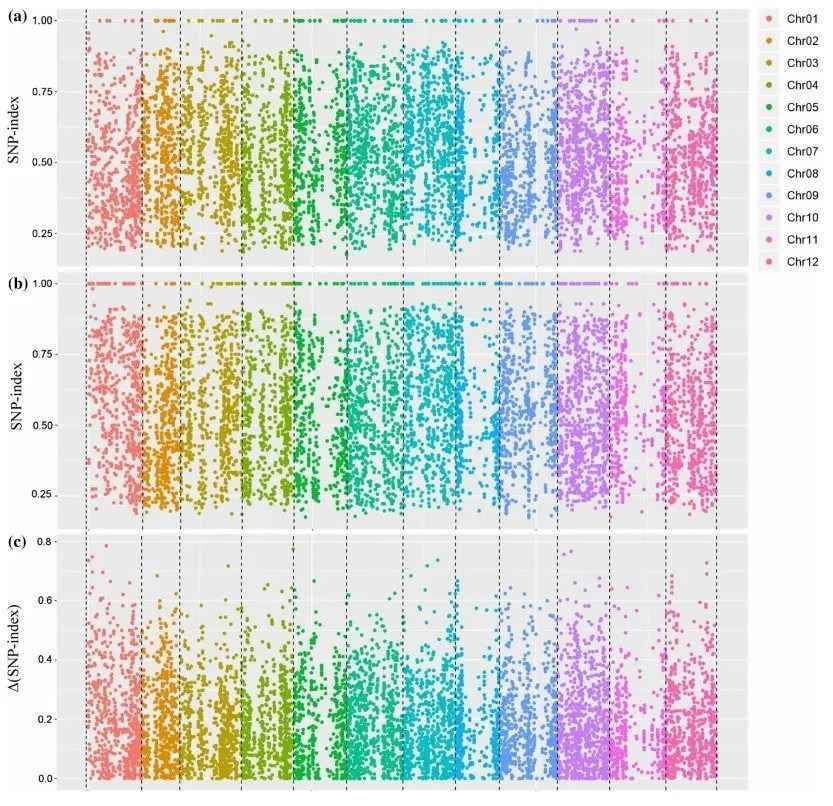 SNP-Index and Δ(SNP-Index) Calculation Results Example
SNP-Index and Δ(SNP-Index) Calculation Results Example
经典案例:基于块状分离分析的下一代测序加速了大豆中两个定性基因的同时鉴定
期刊: 植物科学前沿
影响系数:6.627
发布日期:2017年5月31日
摘要
块状分离物分析(BSA) 为快速鉴定与表型密切相关的分子标记提供了一种简单的方法。BSA技术已被用于各种物种的重要基因定位。随着DNA测序技术的进步,基于BSA的方法 下一代测序(NGS) 显著加快了致病基因的鉴定。大豆是全球种植最广泛的豆科作物之一,由于遗传变异有限、基因组复杂庞大、遗传转化效率低,在基因鉴定和分离方面落后于其他作物。目前只发现了少数控制特定性状的基因,如茎生长习性和每荚种子数。开发快速定位控制重要农艺性状的基因的方法对于推进大豆基因功能研究具有重要意义。
 SNP-Index and Δ(SNP-Index) Calculation Results Example
SNP-Index and Δ(SNP-Index) Calculation Results Example
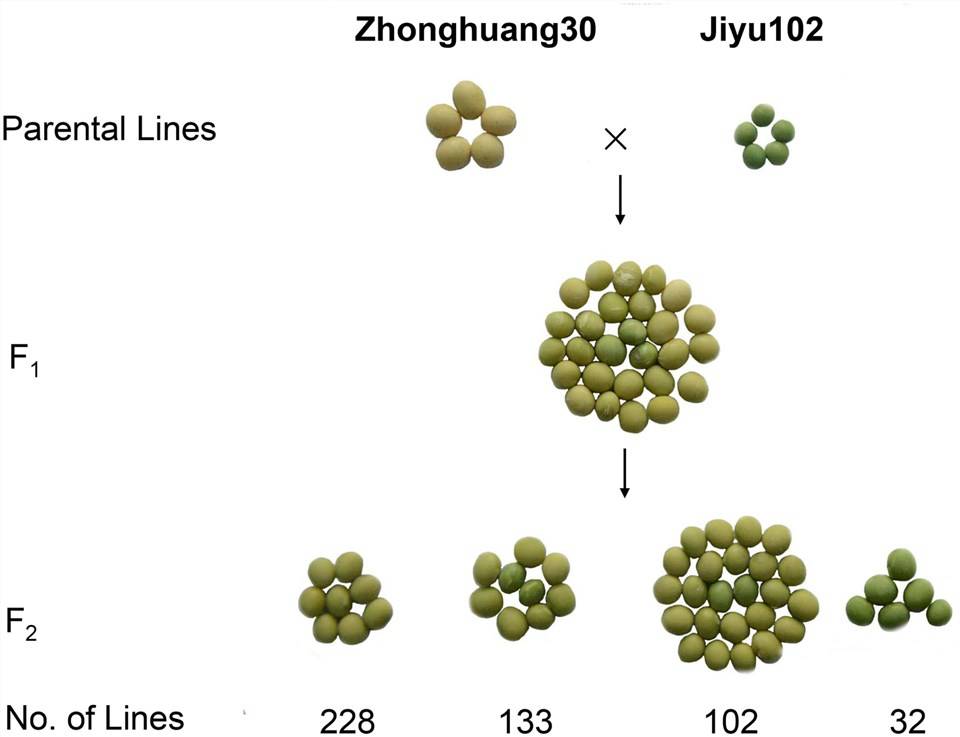
样本来源:F2主页系,专注于黄色/绿色叶色表型
样本量:2个亲本系+30个黄叶后代+30个绿叶后代
测序深度:亲本系(12X+9X),后代(59X+53X)
结果:
中黄30为黄叶亲本,吉育102为绿叶亲本。通过与中黄30(黄叶亲本系)回交F1代,没有发生分离,证实了亲本系中黄色的优势。根据F2代和F2:3主页系的分离率,确定叶色受两个基因控制,绿色是隐性性状。
Using the QTL-seq method, the preliminary localization of the target trait (leaf color) was conducted, and based on ΔSNP-index, the target trait was initially mapped to two intervals, qCC1 (~2.68Mb) on Chr1 and qCC2 (~2.68Mb) on Chr11. Subsequently, fine mapping was performed using SSR and Indel recombination markers on 200 offspring to narrow down the mapping regions. qCC1 was finely mapped to a 30.7kb region, which included four genes, with the D1 gene being a homolog of the green gene in Arabidopsis, as previously reported in other studies. qCC2 was finely mapped to a 67.7kb region, which included nine genes, and similarly, one of these genes, D2, is a reported homolog of the green gene in Arabidopsis.
对这两个候选基因进行了功能探索和验证。发现D1基因在亲本系之间缺少T碱基,导致绿色亲本系JY102发生移码突变和过早终止。此外,D2基因在亲本系之间有一个322bp的序列重复,导致该基因在绿色亲本系JY102中过早终止。
Q1:什么样的亲子关系适合人口结构?
A1:建议选择杂合位点和单性状差异最小的亲本系。此外,亲代之间的差异不应太大,因为过度的分化会导致明显的假阳性,从而难以确定真正的目标区域。在为目标性状选择亲本系时,可以采用各种方法,如EMS诱变、自然个体和紫外线诱变,所有这些方法都可以用于批量分离分析(BSA)。
问题2:选择单个谱系而不是混合种群、自然种群或树木种群的理由是什么?
A2:混合种群、自然种群和树木种群通常具有显著的杂合性,导致大量的遗传多样性。即使在有限的DNA区域内,多个等位基因基因型的共存也很常见。DNA池中不同基因型的流行导致单核苷酸多态性(SNP)检测和基因型频率准确计算的可靠性降低。
在杂交谱系中,一个DNA片段通常只包含两个等位基因,每个等位基因来自亲本,这使得与参考序列的读取比对和变异检测相对简单。在高度杂合的人群中,汇集来自不同来源的DNA增加了读取的多样性,导致参考基因组比对和SNP检测的错误率更高。
In BSA-seq analysis based on the hybrid lineages, most mutant genotypes are high-frequency (≥0.5), making them relatively easy to detect. In contrast, natural populations often exhibit numerous low-frequency SNPs, and when pooled, it becomes difficult to distinguish true low-frequency SNPs from those resulting from sequencing or alignment errors, introducing complexities to data analysis.
Q3:从种子样本中提取亲本DNA,从叶片样本中提取后代DNA是否可行?
A3:选择特定的植物部分进行DNA提取取决于两个关键因素:种皮和胚乳的构成,以及纯合子的程度。主要归因于种皮内母本的起源可能会产生影响。同样,后代的纯合子程度也是一个关键的考虑因素。在纯合性很高,父母和后代DNA之间的差异很小的情况下,这种影响就会减弱。
Q4:汇集后代DNA的推荐程序是什么?
A4:建议从每个后代样本中单独提取DNA,然后以等摩尔量将其合并。这种方法用于最小化背景噪声并减轻潜在的系统误差。
Q5:后代群体应该满足哪些标准?
A5:原则上,任何表现出目标特征的性状分离的杂交后代都可以适用于批量分离分析(BSA)。常用的群体包括F2代、回交群体和重组自交系。对于定性性状,后代可能表现出1:1或3:1的比例,具体取决于所研究的特定性状。在数量性状的背景下,后代表现出符合正态分布的性状是有利的。在明显偏离正常的情况下,必须评估隐性致死基因的潜在影响。
Q6:是否有可能确保候选区域的大小和候选基因的数量?
A6:候选区域的维度和候选基因的数量取决于各种因素,包括种群规模、亲本材料之间的遗传分化程度、目标性状的属性、测序深度和研究物种特有的基因组环境。这些参数可以通过项目特定经验和相关文献进行估算。
Q7:如果映射的区间显示过大,可以采取哪些措施进行调整?
A7:为了优化映射区间,可以调整置信区间,或者选择在BSA映射区间内选择特定的SNP和InDel标记,随后创建局部图。该策略被证明在最小化绘图区域的范围方面是有效的。
Q8:候选基因选择后,如何验证靶基因?
A8:常见的验证方法包括:
SNP验证:
将候选SNP转换为CAPS或dCAPS标记进行验证。
分析候选SNP的限制性酶识别位点,选择引起酶识别位点变化的SNP,使用特异性引物扩增相应的含SNP片段,然后进行酶消化和凝胶电泳,将SNP转化为CAPS标记。分析CAPS标记的多态性以进行验证。
PCR扩增候选SNP区域,并使用Sanger测序验证扩增产物。
使用RT-PCR验证不同表型中的候选基因表达。
基于转录组进行差异基因表达分析,以检查基因表达的显著差异。
RNAi分析:使用RNAi技术特异性沉默或敲除特定基因的表达。
Q9:在人口建设过程中,对父母关系有什么要求?
A9:亲本系应尽可能纯,这种纯度可以通过自花授粉来实现。两个亲本系在目标性状上应表现出显著差异,但其他性状应尽可能一致,以尽量减少后续作图分析中的干扰。
Q10:建议BSA-seq中目标性状的亲本系纯合性背后的基本原理是什么?
A10:BSA-seq性状作图的基本概念依赖于从亲本来源鉴定SNP,并随后计算后代池内整个基因组的SNP指数。如果亲本系不是纯合的,这可能会导致后代SNP的检测率降低和SNP指数降低。因此,为了实现成功的映射,有必要降低SNP指数选择的阈值,这可能会导致假阳性的发生率增加。在计算SNP指数时,目前的做法是使用亲本系作为参考并过滤纯合位点,其中随后在这些特定位点的后代库中计算SNP指数。
Q11:收集后代样本的最佳数量是多少?
A11:后代样本的收集应遵循以下准则:对于定性特征,建议收集尽可能多的隐性个体,至少20个,通常在30到50个之间。同样,应收集同等数量的占主导地位的个体。关于数量性状,通常建议选择表现出最极端性状值的前5%-10%的个体进行进一步评估。此外,基于后代的性状数据创建直方图在指导选择过程方面具有重要价值。

Q12:在重新测序过程中,亲本和后代系所需的测序深度是多少?
A12:为确保SNP和InDel标记的准确性,测序应保持一定的深度。建议亲本系的测序深度不小于20X。合并样本的测序深度应根据样本数量确定,每个样本的平均深度不小于1X。例如,如果有30+30个后代,那么每个后代池的测序深度不应小于30X。如果预算限制允许,您可以考虑进一步增加深度。
Q13:简化的基因组测序可以用于BSA性状作图吗?
A13:缩减基因组BSA技术只能捕获整个基因组的1%到10%。如果所研究的物种具有较大的基因组,并且感兴趣的性状由具有许多相关位点的多个次要基因控制,那么减少的基因组测序可能会捕获一些位点,但会错过其中的大多数,这可能不利于进一步的研究。对于由主基因控制的定性性状或定量性状,存在与基因组BSA减少相关的高风险。建议对亲本系和群体进行简化基因组测序,并构建性状基因图谱的全基因组连锁图。
参考文献
- Michelmore RW、Paran I、Kesseli RV。通过批量分离分析鉴定与抗病基因相关的标记:一种通过分离群体快速检测特定基因组区域标记的方法。 美国国主页科学院院刊. 1991;88(21):9828-9832.doi:10.1073/pnas.88.21.9828
- 张凯,李勇,朱伟,等。黄瓜绿叶基因v-2的精细定位及转录组分析。 前线植物科学。 2020;11:570817.2020年9月25日发布。doi:10.3389/fpls.2020.570817
- Lee SB,Kim JE,Kim HT,Lee GM,Kim BS,Lee JM。基于GBS的BSA-seq对c1基因座的遗传作图揭示了伪反应调节因子2作为控制辣椒果实颜色的候选基因。 应用遗传学理论. 2020;133(6):1897-1910.doi:10.1007/s00122-020-03565-5
- Guo Z,Cai L,Chen Z,等。BSA-Seq和RNA-Seq鉴定孕穗期寒地水稻耐冷性候选基因。 R Soc开放科学. 2020;7(11):201081.2020年11月18日发布。doi:10.1098/rsos.201081
- 赵Z,盛X,余H,王J,沈Y,顾H。菜花抗黄候选基因的鉴定。 国际分子科学杂志. 2020;21(6):1999.2020年3月15日发布。doi:103390/ijms21061999
- Aguado E, García A, Iglesias-Moya J, et al. Mapping a Partial Andromonoecy Locus in Citrullus lanatus Using BSA-Seq and GWAS Approaches. 前沿植物科学. 2020;11:1243.2020年8月19日发布。doi:10.3389/fpls2020.01243
- 宋,李,刘,郭,邱。大体积分离分析的下一代测序加速了大豆中两个定性基因的同时鉴定。 前沿植物科学2017年5月31日;8:919.doi:10.3389/fpls2017.00919。PMID:28620406;PMCID:PMC5449466。