为了正确利用潜在的基因组组装,完整和精确的基因注释非常重要。高效短读RNA测序(或RNA-seq)创新的产生通过促进基因组注释的增强和尚未用于参考基因组的生物体的实验,显著推进了基因组研究。即使如此,无参考和基于参考的短读转录组装也是困难的,并且通常无法对齐经过实验验证的基因模型。事实上,对农业相关植物物种等复杂基因组的注释尚未提供最佳结果。底层基因组组装的不一致性,与不完美的基因模型重建有关,使基因注释变得复杂。
细胞或组织中显示的所有基因都由转录组表示。RNA测序(RNA-Seq)允许识别这些被捕获的基因。对于基因表达差异以及基因型或环境对其表达的影响的实验,创建参考转录组至关重要。通过短读测序,大多数研究生成了一个参考转录组,并通过将读序列组装和/或映射到其他可访问的参考基因组来重建转录组。然而,对于冗长的转录本、重复序列和转座因子来说,这是复杂的。
对于复杂的多倍体基因组,这尤其具有挑战性。最近,以长读测序(LRS)技术为代表的 PacBio 测序和 纳米孔测序,已经可以获得,这项技术通过将全长序列数据创建为单个读取序列来克服这些挑战,包括长转录本(例如大于10kb的转录本),而不需要进一步组装。在一些植物研究案例中,这种方法已被使用,并提供了有关转录差异的额外数据,如选择性剪接和选择性聚腺苷酸化。
太平洋生物科学公司(PacBio)技术(异构测序)提供了最大的测序潜力。它还可以展示全面的数据分析。相反,纳米孔直接RNA测序具有以下优点:(1)与快速、简化的工作流程相结合,最小的功率量允许对基因表达进行高度精细的分析,(2)全长转录物(纳米孔测序传达的长、全长读取的高输出量可以明确描述剪接变异和基因融合),(3)转录物和同工型的精确分类,(4)使用直接RNA测序消除PCR偏差,(5)使用直接核糖核酸区分碱基改变和核苷酸序列,(6)简单识别反义转录物。
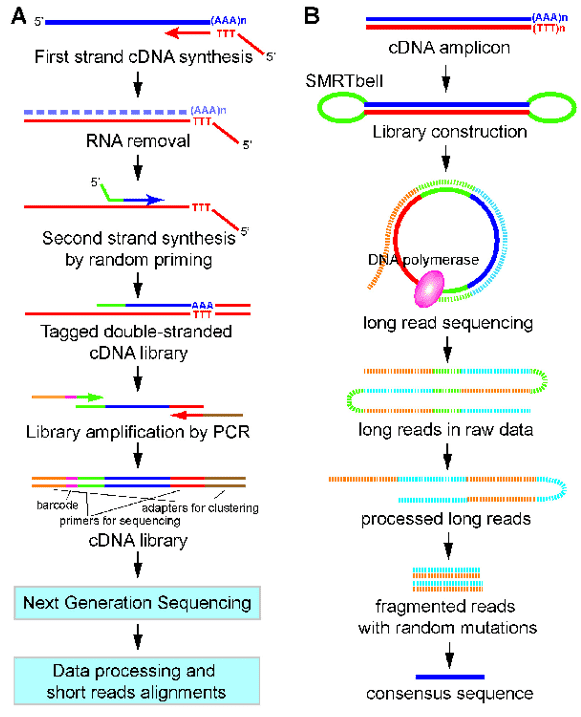
图1。3'end-seq(A)和PacBio Iso-seq(B)的工作流程。(叶,2017)
在功能方面,在医学研究和农业分析领域,PacBio Iso-Seq可以被利用。它可用于医学领域的转录物注释、融合基因探索和疾病机制评估。它可用于功能研究、融合基因探索、推进和压力测试,以及农业领域基因预测和基因组注释的协调。另一方面,Nanopore Direct RNA-Seq可用于评估基因功能,例如聚焦于具有明确功能的样本,以揭示不同功能的主要原因,基因结构,如替代剪接、APA、融合基因、SSR、CDS预测、TSS/TES鉴定,全长转录物量化,如定位广泛有效的差异转录物并识别功能基因,以及RNA甲基化,如直接全长转录组测序,可以在RNA水平上识别碱基改变,如m6A/m5C。

图2:纳米孔直接RNA测序。(Byrne,2017)
参考文献