在早期,微生物的鉴定依赖于表型方法。随着遗传方法的进步,基于核苷酸序列的各种分类方法因其特异性和速度而越来越受欢迎。基于基因测序的技术可以用于检测、鉴定和监测微生物,甚至是鲜为人知或未培养的微生物。
桑格测序
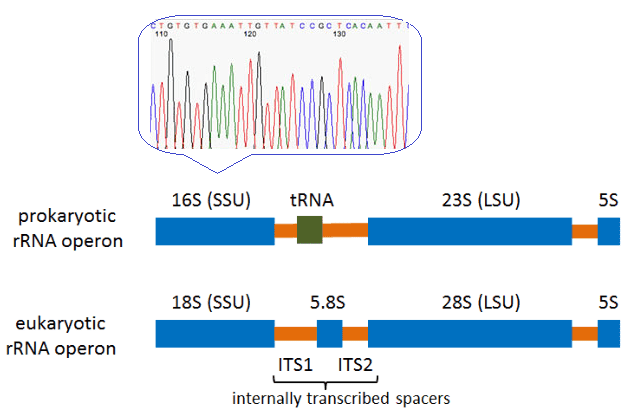
不同生物体基因序列的比较是敏感和精确鉴定微生物的分子工具的基础。目前,核糖体RNA(rRNA)基因序列的直接比较可能是鉴定许多生物体的最有力工具。作为一种补充培养方法,16S rRNA基因测序已成为一种准确、快速的细菌鉴定方法。18S或内部转录间隔基因ITS1和ITS2的测序在真菌分类中起着类似的作用。一个庞大且不断增加的序列数据库允许识别大量微生物。
Sanger sequencing of the rRNA gene is the most widely used molecular tool for microorganisms’ identification. It employs amplification and Sanger- based sequencing of rDNA targets for the identification of bacteria, mycobacteria, and fungi from cultured isolates. The precision of the identification (ie, whether an organism is identified to genus or species level) depends on sequence homology within the database. If it could not be identified to any previously referenced genus, the lack of rDNA sequences for many described species is probably the major cause of failure in molecular identification.
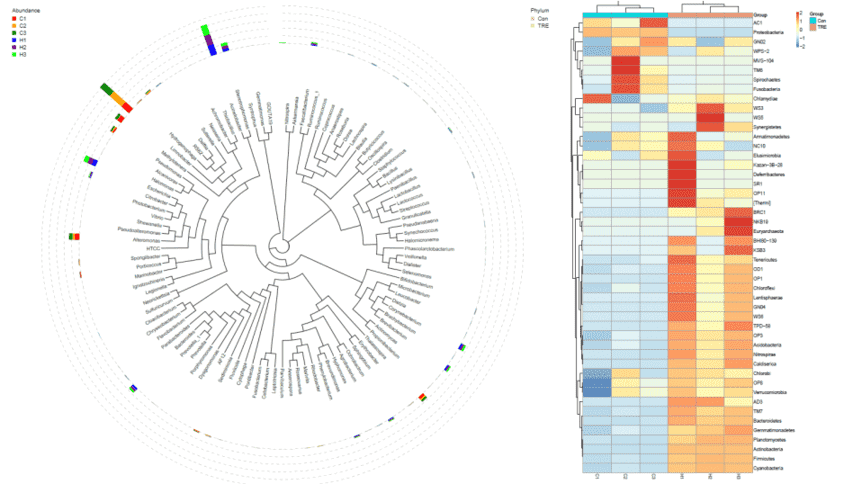
当新序列与数据库中的任何序列之间没有密切匹配时,可以使用分子系统发育的方法对未知微生物进行分类。菌株的系统发育关系来源于所选系统发育标记(如16s rRNA基因)的序列比对。当一个测试生物体聚集在一组属于同一属的细菌中时,通常认为已经获得了属水平的鉴定。
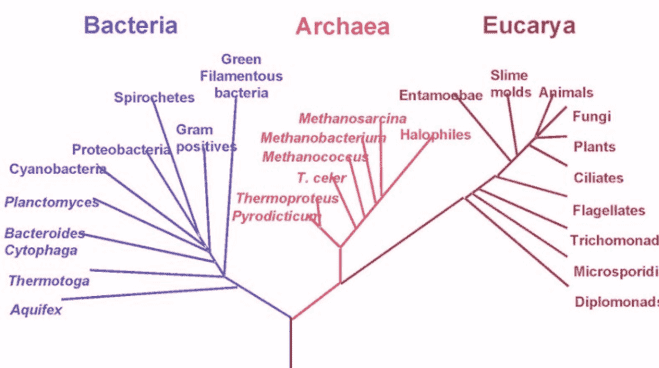
尽管16S rRNA基因测序已成功用于细菌鉴定,但有时两种细菌具有高度相似的16S rRNA序列,因此无法通过该技术区分。在这种情况下,基因组比较可用于确定两个基因组是否来自同一物种。每个物种越来越多的基因组序列的可用性为基于全基因组信息进行距离测定提供了可能性。查询生物的基因组可以与所有已知的细菌基因组进行比较,以在物种水平上识别菌株,为物种定义提供有价值的见解。
桑格测序是一种准确且经济高效的方法,广泛用于细菌鉴定,但它强烈偏向于在实验室环境中易于培养的微生物。由于规模经济和数量级,各种下一代测序平台的测序通量更高,16s/18s/ITS扩增子测序能够对混合微生物群落的集体基因组集进行独立于培养的研究,以并行检测和鉴定几种生物。16s/18s/ITS扩增子测序方法能够分析居住在环境生态位、植物或动物宿主中的群落中的整个微生物群落。
与利用illumina产生的短读V3-V4、V4-V5或V5-V6扩增子的微生物分析研究相比,长读PacBio测序技术提供了全长16S/18S/ITS序列,实现了群落组成和系统发育序列的更深入菌株水平分辨率。基于NGS的16S和ITS rRNA测序方法的另一个优点是,它们提供了一种经济有效的技术来鉴定使用传统方法可能无法发现的菌株。