外显子组测序是一种基因组分析方法,利用序列捕获技术捕获和富集全基因组外显子区域的DNA,然后进行高通量测序。通过仅对整个基因组的约2%进行测序,外显子组测序可以提供超过95%的已知功能区域的信息,包括95%的与孟德尔疾病相关的致病突变和各种疾病易感位点。因此,外显子组测序已成为疾病相关基因检测的常用方法,广泛用于鉴定复杂疾病的致病基因和易感基因。
1) SNP和InDel变异的筛选
SNP和InDel变异根据以下标准进行筛选:未检出率 >= 10%:检出率低于10%的变异被筛选掉;次要等位基因频率 < 0.05:次要等位基因频率低于0.05的变异被筛选掉;非哈迪-温伯格平衡(HWE)位点:不符合HWE的变异,即HWE P值小于0.001,被筛选掉。
应用这些筛选标准后,选择剩余的变异进行进一步的分析和解释。
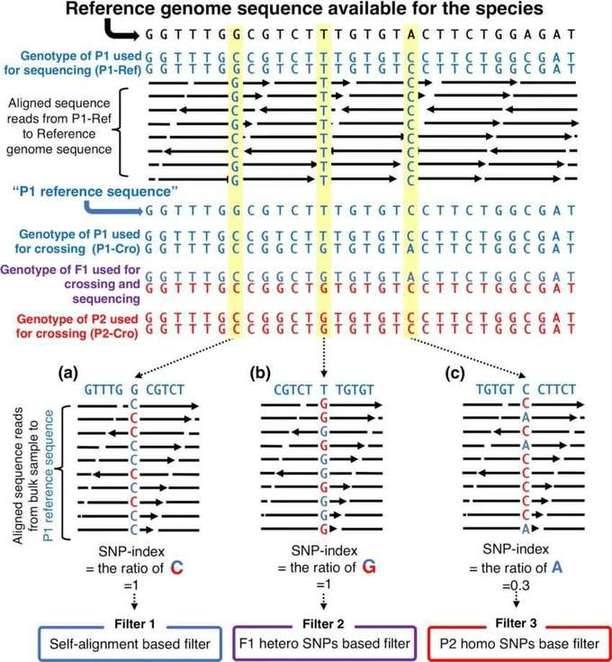 图1:筛选SNP位点
图1:筛选SNP位点
2) 频率计算
频率计算涉及确定病例组和对照组中每个变异的等位基因和基因型频率。
在此步骤中,分别计算病例组和对照组中不同等位基因和基因型的频率。这些频率为研究群体中特定遗传变异的分布和发生提供了有价值的信息。
3) 关联分析
在关联分析中,进行各种统计检验以检查遗传变异与病例对照组之间的关系。
分析包括分层分析、卡方检验、费舍尔精确检验和Cochran-Armitage趋势检验。这些检验用于比较病例组和对照组之间等位基因和基因型的频率分布,并确定是否存在统计学显著差异。
分层分析有助于评估人群特定亚组中遗传变异与疾病风险之间的关联。应用卡方检验、费舍尔精确检验和Cochran-Armitage趋势检验来检查两组之间等位基因和基因型的频率分布是否存在统计学显著差异。这些检验为特定遗传变异与所研究的表型或疾病之间的潜在关联提供了有价值的见解。
4) 单倍型分析
在单倍型分析中,选择包含重要SNP的基因组区域,使用Haploview等单倍型分析软件进行进一步研究。
分析旨在识别和表征这些区域内的常见单倍型。比较病例组和对照组之间已识别单倍型的频率。使用卡方统计量进行此比较,以确定特定单倍型与所研究的疾病或表型之间是否存在显著关联。
通过检查病例组和对照组中不同单倍型的分布和频率,该分析为了解特定单倍型与所研究疾病的潜在相关性提供了见解。使用卡方检验和相应的p值来评估观察到的关联的统计显著性。
 图2:单倍型分析
图2:单倍型分析
5) 变异筛选
在变异筛选过程中,应用了几个标准来选择高质量的候选变异。
首先,获得每个碱基的Phred质量分数,并去除质量分数低于45的候选变异。该阈值确保将测序质量低的变异排除在进一步分析之外。
接下来,检查杂合变异。杂合变异应具有至少10倍的覆盖深度。此外,相应的序列读数应具有不同的起始和结束位置,表明存在等位基因变异。不符合这些标准的变异将被筛选掉。
值得注意的是,人类基因组包含正常的遗传变异,包括单核苷酸多态性(SNP)。为了区分与疾病相关的变异和这些正常变异,利用了dbSNP、1000基因组计划和NHLBI-ESP6500等数据库。与这些数据库中已知的正常变异相匹配的候选变异将被筛选掉,从而降低假阳性率,并专注于潜在的致病变异。
6) 同义变异
这些变异发生在基因的编码区,但不会改变蛋白质的氨基酸序列。就其功能影响而言,它们通常被认为是良性或中性的。
非同义变异:这些变异导致蛋白质氨基酸序列的改变。根据氨基酸变化的性质,它们可以进一步分为错义变异(编码不同的氨基酸)、无义变异(引入过早的终止密码子)或移码变异(阅读框被破坏)。
过早终止变异:这些变异在编码序列中引入过早的终止密码子,导致蛋白质的截短。它们与蛋白质功能的丧失有关,并且可能是致病的。
剪接位点变异:这些变异影响对正常RNA剪接至关重要的剪接位点。剪接的破坏可能导致异常的基因表达和蛋白质产生。
插入缺失:插入缺失是指DNA序列中核苷酸的插入或删除。它们可能导致移码或改变阅读框,从而导致所得蛋白质序列的改变。
通过对这些变异进行分类和分析,可以确定它们的数量和基本信息。这些信息对于理解变异的潜在功能影响及其与疾病或遗传性状的相关性非常有价值。
7) 基因融合
使用FusionMap和GASVPro等工具进行基因融合鉴定,以寻找涉及基因融合的潜在基因组重排。
8) 变异的蛋白质结构预测:使用Swiss-Model工具进行同源建��,以预测变异蛋白质的三维结构。此外,可以使用Chimera或DS等工具进行定点诱变模块分析。建模后,进行动态优化以预测突变对蛋白质结构的影响。
 图3:变异的蛋白质结构预测
图3:变异的蛋白质结构预测
如果获得了少量与所研究表型相关的基因变异,则进行文献综述和数据库检索(如ClinVar、COSMIC数据库)以获得支持实验结果的潜在相关基因功能。如果获得了大量相关基因,则选择GO分析来验证突变基因是否富集于与表型特别相关的功能。
步骤1:从体细胞突变中优先选择候选癌症驱动突变。
为了评估罕见和常见突变对癌症发展的影响,我们首先采用径向支持向量机(SVM),该机器在来自COSMIC和UniProt数据库的非同义单核苷酸变异(nsSNV)上进行训练。使用体细胞突变作为输入,确定系统计算相应的径向SVM分数,以预测每个突变成为癌症驱动因素的潜力。
步骤2:从候选驱动突变中识别候选驱动基因。
基于数十年来从研究中产生的宝贵知识,我们在径向SVM层之上增加了另一层。该层根据每个突变对应基因的Phenolyzer分数来衡量其权重,从而根据先前的知识评估每个突变基因型和表型之间的遗传关联。然后根据其总加权分数对包含有害突变的基因进行筛选和排序。
步骤3:从候选驱动基因中识别候选药物。
为了更好地协助研究人员/临床研究人员进行潜在的个性化治疗,我们在确定过程中引入了第三步,为每个癌症基因驱动因素提供优先的药物目录。此步骤根据候选药物与我们预测的癌症基因驱动因素的相互作用来识别候选药物,并使用相应的靶基因相互作用分数为其加权。通过查询PubChem数据库获得药物活性分数。
使用RNAsnp软件评估单核苷酸变异(SNV)对RNA二级结构的影响。结构评估如下:
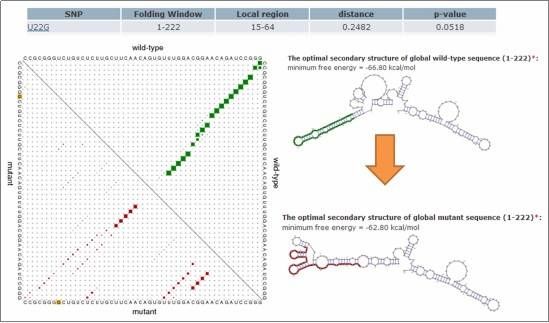 图4:SNV对RNA二级结构的影响
图4:SNV对RNA二级结构的影响
通过使用ENCODE数据库的组合分析来确定转录调控因子与目标SNV的关联。该分析有助于识别其结合受SNV影响的转录因子。
 图5:与SNV对应位置的转录因子
图5:与SNV对应位置的转录因子
使用我们内部开发的网络分析系统RBP-Var分析目标SNV对转录后调控的影响。该系统评估SNV对转录后调控的影响,并识别潜在的调控机制。
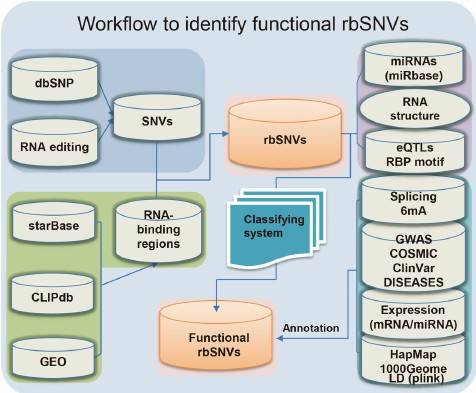 图6:SNV转录后调控网络分析系统
图6:SNV转录后调控网络分析系统
我们利用dSysMap:探索疾病突变的边缘作用来构建癌症基因相互作用网络。该工具使我们能够探��疾病突变在癌症背景下对蛋白质-蛋白质相互作用的功能影响。
通过利用预测软件,我们可以评估目标SNV对蛋白质三级结构的影响。该分析揭示了与野生型基因相比,有害突变引起的蛋白质结构变化。此外,它还使我们能够研究突变蛋白质对蛋白质-蛋白质相互作用的影响。此外,它还能够研究突变对蛋白质相互作用的影响。
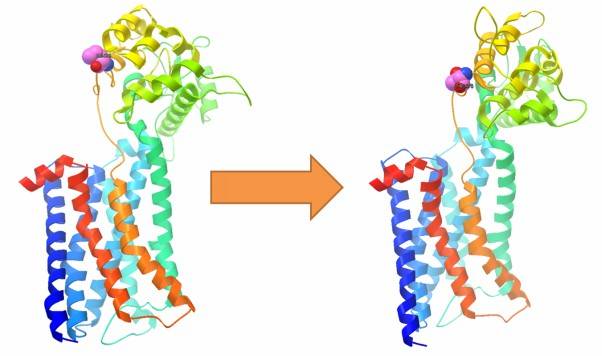 图7:SNV对蛋白质三级结构的影响。
图7:SNV对蛋白质三级结构的影响。
图8:SNV对蛋白质-蛋白质相互作用的影响。
参考文献: